Close
Section of Pharmacogenetics – MIS-lab
Our research aims at elucidate the bases for interindividual differences between individuals in liver function, risks for liver diseases, drug response and risk for adverse drug reactions as well as to identify new drug targets for treatment of steatosis and liver fibrosis, using a novel in vivo-like 3D liver spheroid system.
Part of the:

Latest news
MIS-lab
Our research aims at understanding the basis for liver diseases, drug induced injuries and the differences between individuals in liver function, risk for liver diseases, liver regeneration, human drug metabolism, drug toxicity and drug response, thereby facilitating a more effective personalized drug treatment at the clinics.
Background
The liver is a vital organ for synthesis and detoxification. It has an ability to regenerate after partial hepatectomy and other major injuries, however the mechanisms behind are relatively unknown.
If major liver disease occurs transplantation is today the only manner for cure. The most significant liver diseases are:
- hepatitis, inflammation of the liver, mainly induced by various viruses but also drugs alcoholic liver disease including fatty liver disease
- alcoholic hepatitis, and cirrhosis, the formation of fibrous tissue in the place of liver cells that have died
- non-alcoholic fatty liver disease (NAFLD), where hepatic steatosis, accumulation of lipids within hepatocytes, may lead to inflammatory disease (i.e. steatohepatitis) and cirrhosis
- non-alcoholic fatty liver steatohepatitis (NASH) and v) cirrhosis, where the developing fibrosis can cause chronic liver failure. NAFLD constitutes a clinicopathological condition that encompasses a spectrum of liver disorders.
NAFLD is the most common liver disease affecting between 20% and 44% of European adults and 43-70% of patients with type 2 diabetes (Blachier et al, 2013). Onset of NAFLD is hallmarked by the accumulation of lipids within hepatocytes (hepatic steatosis), which arises from an imbalance between triglyceride import, production and extrusion primarily caused by obesity and a hypercaloric diet. Furthermore, steatosis can be induced by a variety of drugs. Steatosis can progress into non-alcoholic steatohepatitis (NASH), an inflammatory condition that can result in liver failure. An additional and important cause of liver injury is adverse drug reactions (ADRs). It is estimated that ADRs cost as much as drug treatment itself, that it causes withdrawal of 4 % of new chemical entities from the market and is the 4th- 5th leading cause of death (cf. Eichelbaum et al., 2006). It has been estimated that adverse drug reactions cause up to 7 % of all hospital admissions in the UK and 13% of all admissions to internal medicine clinics in Sweden are due to ADRs (Cf. Sim and Ingelman-Sundberg, 2011).
Our research aims at understanding the basis for these liver diseases, drug induced injuries and the differences between individuals in liver function, risk for liver diseases, liver regeneration, human drug metabolism, drug toxicity and drug response, thereby facilitating a more effective personalized drug treatment at the clinics. The research encompasses:
- Identification and functional validation of genetic polymorphism of the genes encoding drug transporters, drug metabolizing enzymes and drug targets.
- Implementation of pharmacogenetics into psychiatry by the identification of novel genetic variants of importance for metabolism of CNS drugs and validation in large clinical cohorts.
- Identification of novel mechanisms for drug induced liver injury using 3D spheroid models.
- Identification of the role of different types of liver cells, signal transduction systems and mediators for development of NASH using 3D liver spheroids.
Projects
Mimicking liver function and liver disease in vitro for drug development
1.1 Summary
We use 3D hepatic spheroid systems obtained from human liver donations or cryopreserved hepatocyte cultures in combination with different types of non-parenchymal cells to:
- Evaluate the usefulness of these spheroid systems for studying properties and functions of diseased liver models and their potential for use during drug development including validation of existing drugs and development of new drugs.
- Evaluate in detail mechanisms of formations and treatment of NAFLD, NASH and fibrosis in the spheroids systems including screening for novel drug therapies using the spheroids as validation system.
- Evaluate chronic drug toxicity in diseased liver systems as well as novel mechanisms for drug induced hepatocyte proliferation and cytoskeletal changes.
This information will be of importance for:
- Increased possibilities to avoid the development of hepatotoxic drugs.
- Understanding different liver diseases and
- Validation of drug targets and drug candidates aimed for treatment of liver diseases.
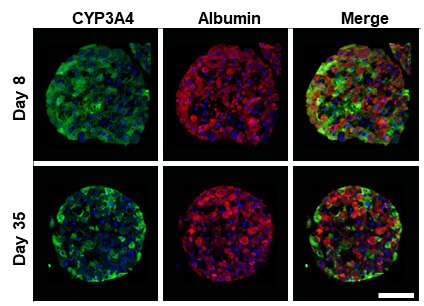
Fig 1. Liver spheroids after day 8 and day 35 in culture stained for CYP3A4 (perivenous) and Albumin (periportal).
1.2 Experimental platform
In our lab we have established and further developed novel spheroid based 3D systems for studies of liver function and properties in vitro (Gunness et al., 2013; Bell CC et al., 2016).
Starting with hepatocytes from commercial sources, we form spheroids from primary human hepatocyte fractions (PHH spheroids) using the ultra-low attachment plates and defined media. In the spheroids, the cells have relevant cell-cell interactions, a physiological cell-extracellular matrix (ECM) interface, improved cell polarity and a functional bile canaliculi network (see Figure 1). Indeed we showed that relevant enzyme expression can be preserved for at least 5 weeks in culture and that periportal and perivenous hepatocytes retain their phenotype during this period (Bell et al., 2016). The hepatocytes in 3D spheroids exhibit molecular phenotypes on transcriptomic, proteomic and metabolomic level similar to the livers from which they originate. We can transfect the spheroids with virus mimicking viral induced hepatitis or supplement the media with bile acids, allowing the faithful identification of drugs with cholestatic liabilities (Hendriks et al., 2016).
Moreover, we can induce steatosis by drugs or by altering the media composition and we can grow the spheroids in the presence of non-parenchymal cells and induce inflammation, hepatitis and liver fibrosis (Hurrell et al., 2020). We have also developed a model for detection of cholestatic drugs.
1.3 Pathological liver systems
Using these diseased liver systems we elucidate the drug hepatotoxicity including their mechanisms (Hendriks et al., 2019) in diseased liver as compared to healthy liver. We also evaluate novel mechanisms for drug mediated enzyme induction (Hendriks et al., 2020), study factors causing steatosis, and fibrosis in the liver and new regimens to inhibit such development.
The transition from steatosis to NASH, fibrosis and HCC is characterized by an intricate interplay of a multitude of nutritional and genetic factors, pathways and cell types. We validate the utility of the 3D spheroid culture system in which PHH can be co-cultured with Kupffer and stellate cells in 96-well format (Bell et al., 2016; Hurrell et al., 2020) as an in vitro disease model for human NAFLD compatible with high-throughput analyses.
We develop culture conditions that closely mimic the nutritional and hormonal status of hepatocytes in vivo with regards to e.g. glucose and glucagon levels, fatty acids and insulin. In this system, we extensively characterize 3D cultured hepatocytes with regards to expression levels and activities of gene products involved in liver homeostasis and causing insulin resistance and metabolic diseases. We have previously shown that our human 3D culture system can recapitulate lipid accumulation, when exposed to elevated levels of fatty acids, fructose and insulin, as is the case in NAFLD in vivo (Figure 2).
Currently we strive hard to develop different models for liver fibrosis and evaluate drug candidates for their treatment.
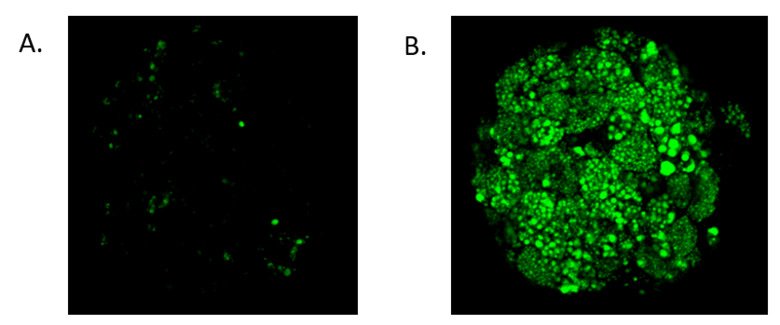
Fig 2. Induction of steatosis using 2 x plasma concentration of FFA and induction of steatosis in the spheroids under metabolic syndrome conditions. A. Control conditions; B. steatotic conditions.
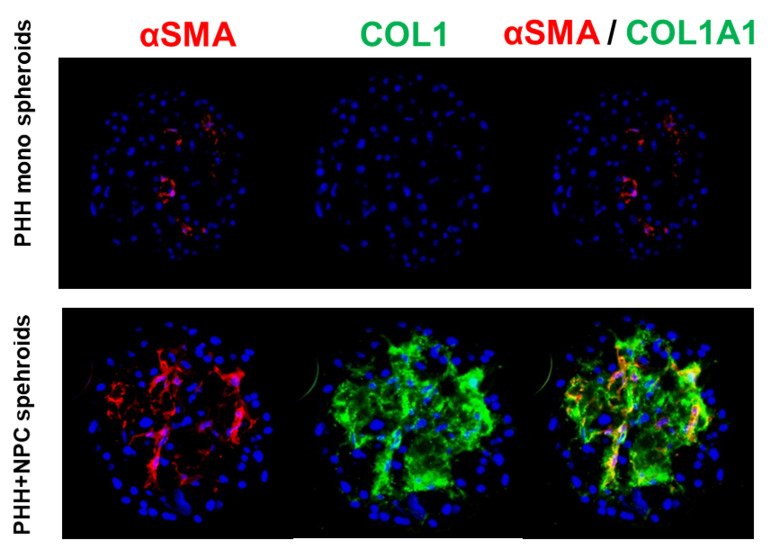
Fig 3. Induction of fibrosis as monitored by collagen 1A1 and smooth actin expression using 2 x FFA concentrations in liver spheroids composed of hepatocytes only or hepatocytes with non parenchymal cells. (From Hurrell et al., 2020)
1.4 Mechanisms for drug induced enzyme induction
In vivo in man drugs can cause induction of enzymes participating in their metabolism. Such induction can lead to resistance for drug treatment and it is important to evaluate preclinically that drug candidates do not harbour such a property. On the basis of a pharmacokinetic failure in clinical trials where animal models and conventional 2D hepatocyte models were unable to predict drug induced induction in vivo, we are, based on the 3D spheroid model, investigating novel mechanisms for drug dependent CYP induction which involves intracellular signal transduction systems. The 3D spheroid system is validated as a new standard in vitro system for prediction of drug mediated enzyme induction.
2. Role of CYP2C19 in brain phenotypes
The CYP2C19 enzyme metabolizes endogenous and synthetic psychoactive compounds including steroid hormones, cannabinoids, SSRIs, tricyclic antidepressants, and benzodiazepines. Two major variant alleles are known, CYP2C19*2 is defective and CYP2C19*17, which is identified by our group, is causing increased enzyme expression.
This project aims to reveal the role of CYP2C19 enzyme in prenatal brain development, as well as its role in adulthood in affective disorders and metabolism of psychopharmaceuticals.
2.1. Involvement of endogenous CYP2C19 in depressive phenotypes and hippocampal homeostasis
We detected CYP2C19 expression in fetal human brain and in collaboration with Nancy Pedersen, we previously showed that the presence of two defective (CYP2C19*2) alleles causes a decrease in depressive symptoms. We found that transgenic mice containing human CYP2C19 gene also show expression of this gene in developing brain, as well as decreased hippocampal volume, anxious phenotype, and increased hippocampal activation after acute stress. In humans, we found that the absence of CYP2C19 is associated with a bilateral hippocampal volume increase in two independent healthy cohorts and a lower prevalence of major depressive disorder and depression severity in African-Americans (N=3,848) (Jukic et al., 2016, 2017). Transgenic 2C19 mice showed high stress sensitivity, impaired hippocampal Bdnf homeostasis in stress, and more despair-like behavior in the forced swim test (FST). So indeed several of the phenotypes originally described to be caused by overexpression of CYP2C19 in mice were also found to be influenced by CYP2C19 in humans which indicates that elevated CYP2C19 expression is associated with depressive symptoms, reduced hippocampal volume and impairment of hippocampal serotonin and BDNF homeostasis.
2.2. Establishing of CYP2C19 genotype-driven dosing regimen of escitalopram
Well replicated in vitro and clinical studies indicate that escitalopram (ESC) is the most selective and most efficient among selective serotonin reuptake inhibitors (SSRIs). SSRIs are the cornerstone of the modern antidepressant pharmacotherapy; however, a large proportion of depressed patients do not respond adequately to the treatment with these drugs. The variation in the disposition of ESC due to CYP2C19 polymorphism may significantly contribute to the inter-individual variability in antidepressant response among the patients. In this project, we found a large contribution of CYP2C19 polymorphism to the escitalopram levels as well as the treatment success based on successful drug utilization as measured by the extent of drug switching (Fig 3).
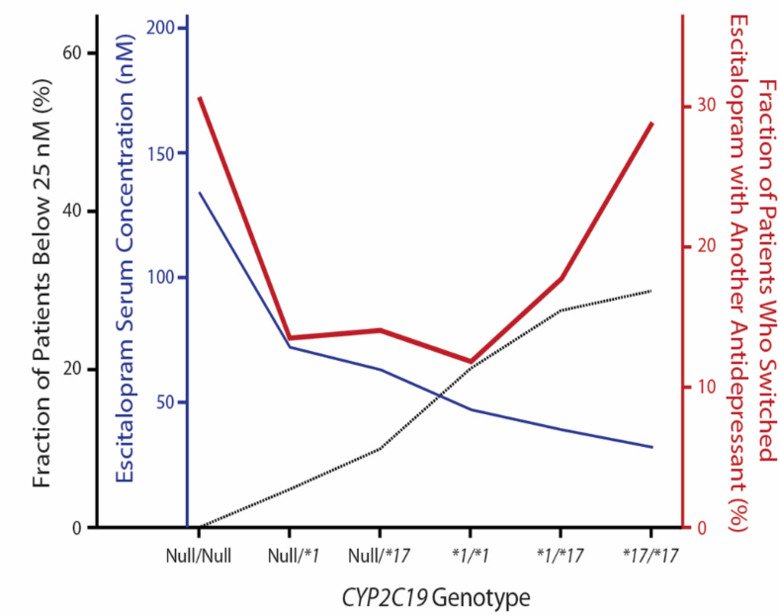
Fig 3. Plasma levels and treatment success using the antidepressant escitalopram in relation to the CYP2C19 genotype. Subjects with higher or lower enzyme capacity get erroneous plasma levels of the drug and have a much higher risk for drug switching.
2.3. Involvement of CYP2C19 in developing brain in physiology and degradation of dopaminergic neurons
Movement disorders, including Parkinson’s disease, significantly contribute to the morbidity caused by diseases worldwide; however their etiology and pathophysiology are still largely unclear. Animals overexpressing the human CYP2C19 gene (2C19TG) exhibit an increased dopamine (DA) concentration in the brain and hyperkinesia in the young age and degeneration of dopaminergic neurons in the old age. Conversely, healthy human subjects of genotype connected with elevated CYP2C19 expression show lower grey matter scores in substantia nigra (SN), indicative of DA neuronal degradation in this brain region. The aims of this project are to: (1) Track the state and function of DA neurons in SN of 2C19TG mice to understand the succession of their early hyperactivity and late (potential) degradation in a systematic manner, (2) Elucidate the alteration in biochemical cascades behind the reduction in dopaminergic neurons in 2C19TG mutants, and (3) Validate the clinical significance of these findings by using neuroimaging techniques on healthy individuals. Overall, the investigation through which CYP2C19 affects dopaminergic neuronal physiology and degradation is aimed to lead to the establishment of genetic and neuroimaging biomarkers, as well as to provide novel drug-targets for the treatment of dopamine-mediated motoric deficits.
3. Evaluation of the importance of rare genetic variants and novel haplotypes on hepatic metabolism and drug response
Genetic variants primarily encoding drug and metabolite transporters, phase I and phase II drug metabolizing enzymes and nuclear receptors can influence drug response by modulating drug absorption, distribution, metabolism and excretion (ADME). Importantly, while in the past decades an ever-growing arsenal of genetic variants with demonstrated impacts on human drug response has been identified in these pharmacogenes, a substantial fraction of the heritable variability in drug response remains unexplained. Rare genetic variants that only occur in very few individuals and are hence missed in genome-wide association studies have been proposed to contribute to this missing heritability. We have recently highlighted the role of CYP2D6 and CYP2C19 polymorphism for better treatment of schizophrenia and depression, respectively. The results indicate important effects on the clinical outcome by personally designing the drug dose based on the genetic variants of these enzymes the patients carry (Jukic et al, 2019; Jukic et al. 2018; Jukic et al., 2021; Milosavljevic et al., 2021).
In collaboration with Espen Moldens group in Oslo and Lili Milanis group at the Estonian Genome Center, Tartu, we currently identify novel genetic loci of high importance for personalization of drug treatment using large patient cohorts. The polymorphisms identified are evaluated in our hepatic spheroid system using gene silencing by e.g. siRNA constructs. We have recently found novel genetic loci for determining the metabolism of antidepressants and antipsychotics as well as novel haplotypes of CYP genes determining drug metabolism (Bråten et al., 2021). These results will allow the construction of novel microarrays for personalized drug treatment in the clinic.
This project is linked to our participation in the H2020 project Ubiquitous pharmacogenomics (U-PGx; http://upgx.eu/) in which we have the primary responsibility to analyze the genetic background of outlier patients with abnormal drug response.
Staff and contact
Group leader
- Magnus Ingelman-SundbergProfessor, Senior
All members of the group
- Andrea AtanasovAffiliated to Research
- Inger JohanssonSenior Lecturer
- Marin JukicSenior Research Specialist
- Åsa NordlingLaboratory Assistent
- Aileen TranAffiliated to Research
- Arnau Vila BertomeuAffiliated to Research
- Sander van RietResearch Specialist
Visiting address
Karolinska Institutet, Biomedicum, Quarter B5, Solnavägen 9, Stockholm, 171 65, Sweden
Postal address
Karolinska Institutet, Department of Physiology and Pharmacology, Ingelman-Sundberg group - Pharmacogenetics, att: (name of recipient), Stockholm, 171 71, Sweden