Research focus
Accumulating evidence suggests that the etiopathogenesis of neurodevelopmental disorders can be linked to specific genetic and environmental factors alone or in combination.
Our studies have focused on the neurodevelopmental alterations caused by different environmental neurotoxic contaminants, such as heavy metals or endocrine disruptors at levels relevant to human exposure, as well as on the detrimental effects of excess glucocorticoids (GC) leading to fetal growth restriction.
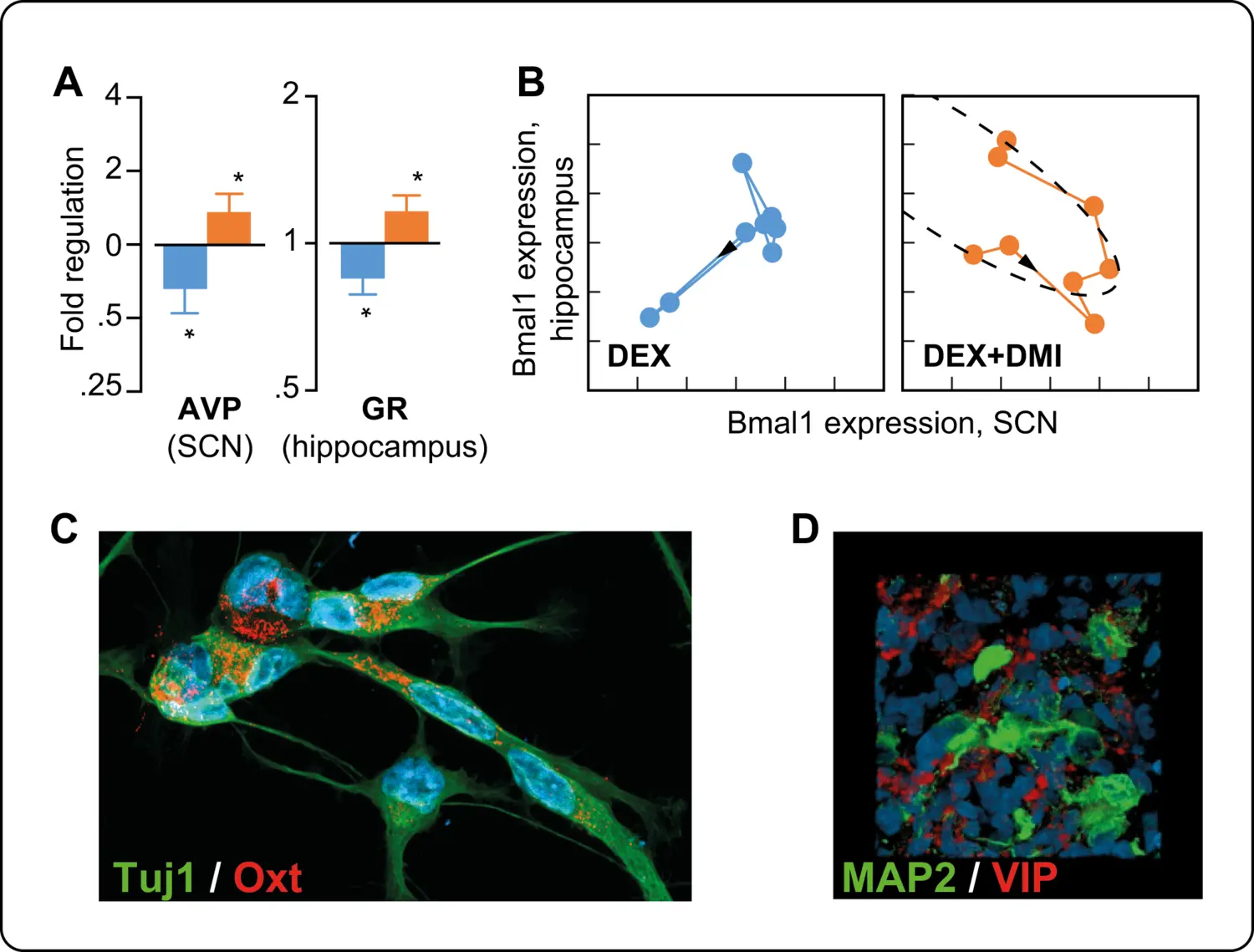
By combining in vitro and in vivo experimental approaches, we could show that high levels of GC induce epigenetic modifications that affect differentiation, and migration as well as mitochondrial function and redox state in neural stem cells. The analysis of behaviour revealed that prenatal excess GC induces late onset of depression-like behaviour in adult male mice exposed to GC in utero associated to decreased hippocampal neurogenesis that could be restored by desipramine but not fluoxetine. Long-lasting alterations in circadian patterns of activity and weaker coupling between the central clock (i.e. suprachiasmatic nucleus) and peripheral oscillators appeared long before the onset of depression suggesting that these alterations may predict the onset of depression and the response to therapy in depressed patients. Our collaborative clinical investigations provides preliminary evidence supporting this hypothesis.
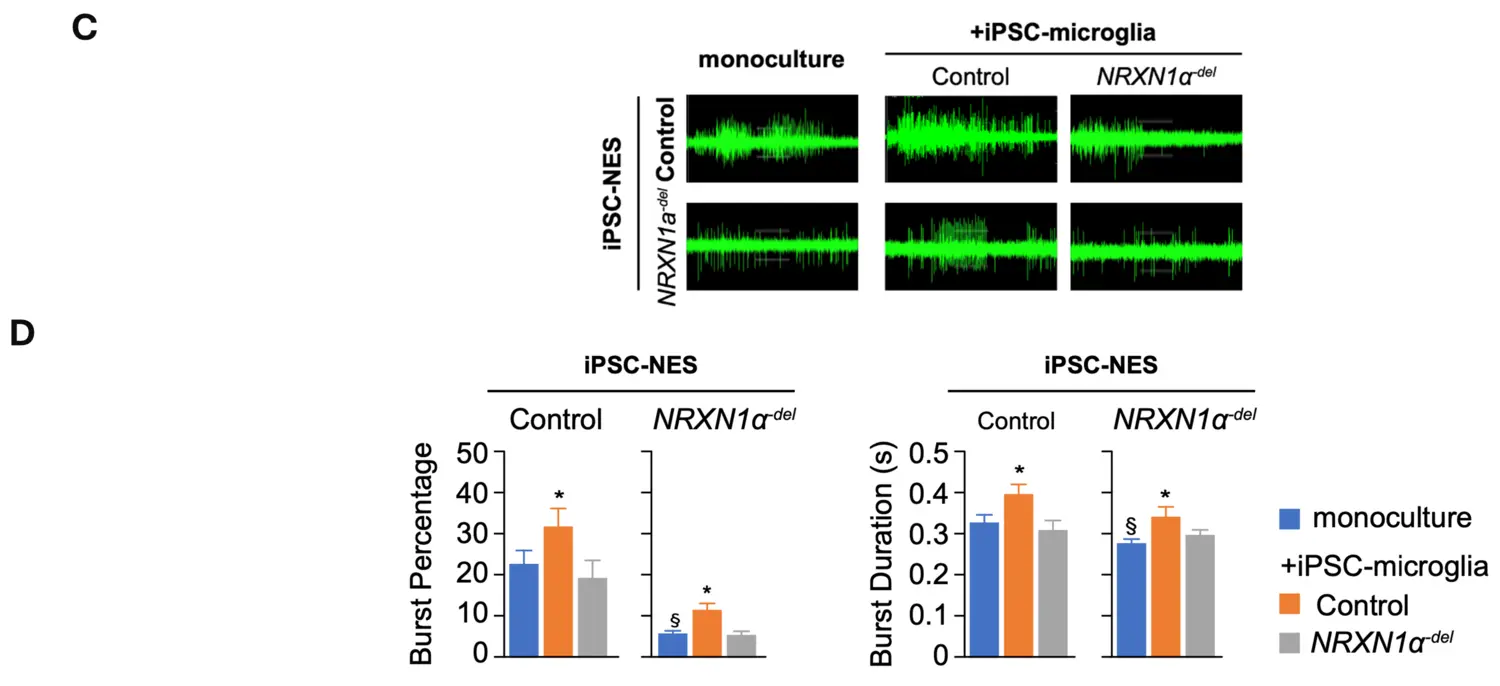
More recently, we expanded our studies on the neurodevelopmental effects induced by the deletion of NRXN1. Specifically, we investigated the ability of microglia to support neuronal circuit formation and found dramatic difference in synchronization across the network in differentiated NRXN1α-del iPSC-NES as compared to control iPSC-NES (figure upper part). Quantification of synchronized burst activity in differentiated iPSC-NES showed that network synchronization is reduced in differentiated NRXN1α-del iPSC-NES as compared to control iPSC-NES in monoculture. The addition of control iPSC-microglia promotes synchronization across neuronal networks in both control and NRXN1α-del iPSC-NES. In contrast, the ability to support network activity is lost in NRXN1α-del iPSC-microglia, as illustrated by the lack of effect in control iPSC-NES, and the negative effect in NRXN1α-del iPSC-NES. Interestingly, the addition of control iPSC-derived microglia to cultures of NRXN1α-del iPSC-NES-derived neurons increased synchronization of neuronal activity. Our data suggest that, in addition to neurons, microglia are also negatively affected by NRXN1α-deletion, and this significantly contributes to the observed neuronal circuit aberrations. Moreover, our data highlighted a direct contribution of IL6 to the observed neuronal phenotype.