Technology development
Cell types in healthy tissues only take on a finite number of states, since they are generated following a strict developmental program, and characterization of every cell type in human tissues is therefore possible. However, cancer cells carry genetic alterations that may remove such constraints, leading to a fundamental challenge: the potential co-existence of genetically distinct clones, each supporting multiple stable cancer cell states. This motivates the development of multiomics approaches for applications in cancer.
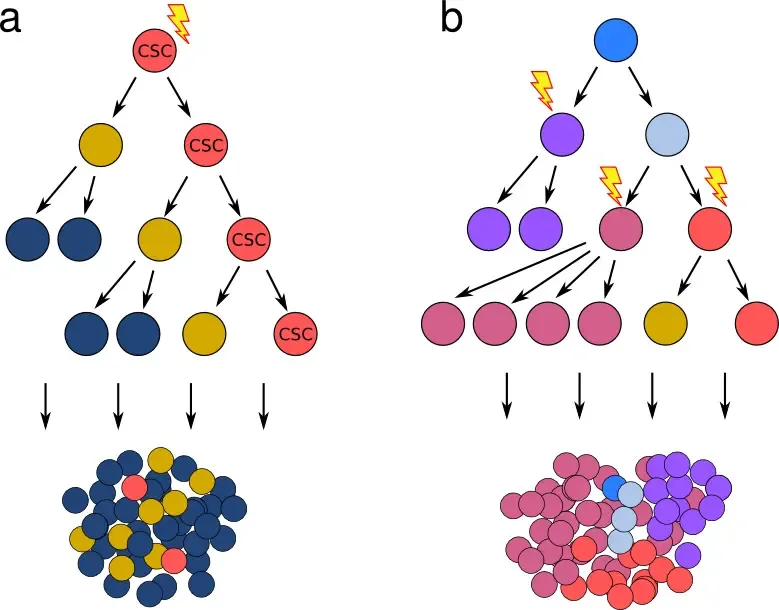
Cancer stem cell vs stochastic model of tumor growth
a) The cancer stem cell hypothesis postulates the existence of rare cells that show stem cell properties (marked CST), and that give rise to genetically identical but phenotypically and epigenetically distinct progeny - resulting in a hierarchy mimicking the differentiation steps in healthy tissue. b) Classical model, where cancer cell characteristics are changed exclusively by a gradual accumulation of stochastic genetic events (mutation, loss/duplication/translocation). Importantly, both models can give rise to a tumor mass with similar characteristics, including fraction of cells with a certain phenotype. Obtaining transcriptomic and genotypic data in parallel is necessary to disentangle which features originate in genetic alterations (b) and which stem from epigenetic changes (a).
Clonal evolution and cell types in cancer
Pediatric Acute Lymphoblastic Leukemia (ALL) arises from lymphocyte progenitors and is known to maintain a hierarchy of cell differentiation, making it a suitable model disease for studying cancer stem cells. Although cure rates for ALL are currently above 80%, the prognosis of patients with relapsed ALL is dismal, with an overall survival rate of only 30%. We are analyzing primary and relapsed ALL from the same patient, with the aim of determining the clonal structure of the leukemia and characterize the cancer stem cell population in the primary sample of patients which later relapse. To differentiate between genetic and epigenetic changes, we are using a novel method that allows us to obtain genotypic and transcriptomic data from the same single cell, allowing us to trace clonal expansions that are mainly driven by epigenetic factors as well as those driven by genetic alterations.
External web site: www.engelab.org