Examples include transcription factors (TFs), closely related but distinct alternative promoters resulting in the same protein but employing different sets of regulatory TFs, expression of anti-sense RNA to modulate the sense-RNA and the regulatory role of enhancers, expressed repeat elements and miRNAs. We are also involved in several genome annotation consortia.
Development of sequencing technologies and sequencing library methods for genome, metagenome, transcriptome and epigenome data is moving at a breathtaking pace. We are working with the development of corresponding bioinformatics data analysis technologies for these genomics data. For example, our group identified gene enhancers in transcriptome data and assigned gene regulatory roles to these enhancers in diseases and development.
Understanding the molecular basis of perturbed gene regulation in diseases is one aspect of our research. We are using genome-wide analysis technologies based on high-throughput sequencing extensively. Developing the necessary bioinformatics tools together with best-practice analysis methods constitutes an important aspect.
For the last ten years, we have been working on obesity-related type 2 diabetes and on asthma. Close collaboration with clinical research groups has been of key importance. We usually join the projects during the design phase where we contribute to the experimental design in terms of cohort stratification, statistical power, selection of tissue types, annotation of samples and data as well as selecting the high-throughput analysis technologies used. Addressing the specific biomedical questions of the projects by analysing the sequencing data together with the clinical data constitutes one of the main aspects of our work. Very close interaction with the clinicians is of outmost importance in connecting the findings of the data analysis to the biology underlying the disease.
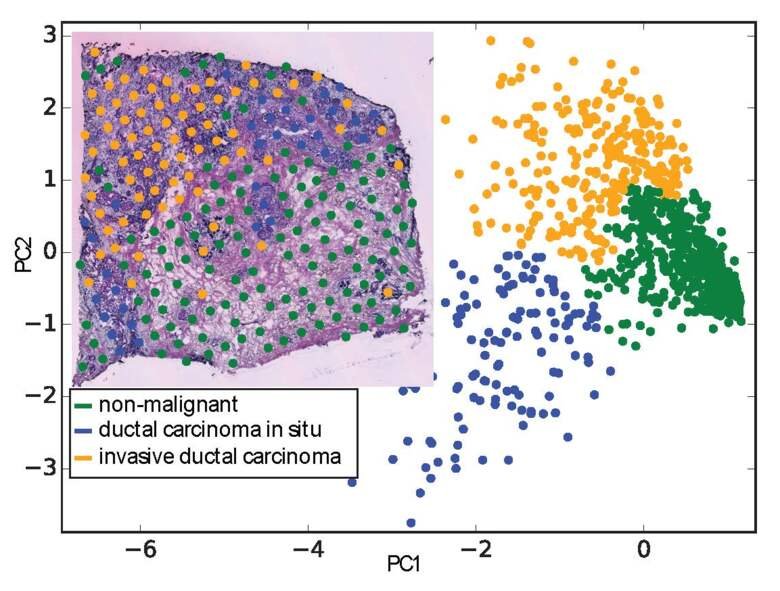
We contributed to genome annotation projects for the zebrafish as part of the DANIO-CODE consortium and for the dog as part of the DoGA consortium. We first developed a sample and data annotation framework since both consortia are consolidating data from various sources. Main goals include improved genome annotation and adding gene enhancers. We work with Spatial Transcriptomics (ST) data, where we used the ST data in a gene independent way and employed machine learning methods to identify breast cancer signatures.
 Photo: SciLifeLab
Photo: SciLifeLabOur research group is a part of SciLifeLab
SciLifeLab is a national collaborative initiative involving multiple Swedish universities, with partnerships across healthcare, industry, government agencies and international organisations. It is a state-of-the-art research infrastructure and environment for molecular biosciences, providing advanced technologies and expertise for both fundamental and applied research in the life sciences.