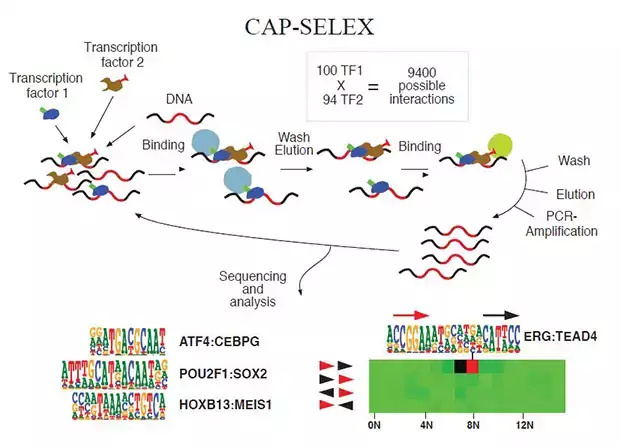
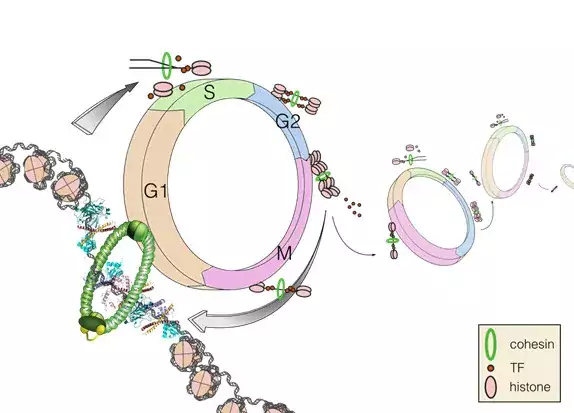
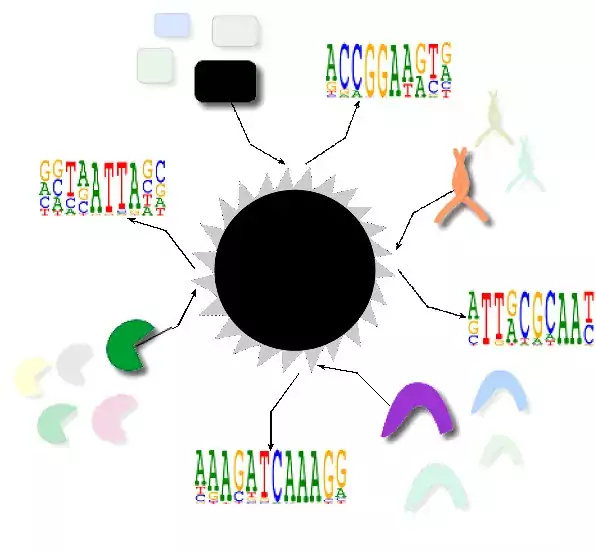
Research
TFs are analyzed both alone, and in combination with other TFs and scaffolding proteins such as the mediator complex. The resulting knowledge is then applied to the interpretation of large data sets such as whole cancer genomes, and genome-wide association studies that have revealed genomic regions associated with a wide variety of diseases, including heart disease, diabetes and different types of cancer. The work in the laboratory is interdisciplinary, and has impact both to basic scientific understanding of gene regulation, and to mechanisms of formation of cancer and other diseases.
The specific objectives of our research are the following:
- To identify mechanisms that govern transcription factor binding in vitro and in live cells
- To use the resulting information in the interpretation of cancer genomes and genome wide-association studies
- To validate the findings in mouse genetic models
About the laboratory
Professor Jussi Taipale got his Ph.D. from the University of Helsinki in 1996, and continued with postdoctoral work at the University of Helsinki and at Johns Hopkins University (Baltimore, MD, USA). He has headed an independent research laboratory since 2003, focusing on systems biology of growth control and cancer. The main expertise of the Taipale group is high-throughput screening using cDNA and RNA interference, computational and experimental methods to identify causative regulatory mutations in non-protein coding DNA and to analyze genetic networks. In addition, Taipale group has extensive expertise on mouse models of gene and regulatory region function. The group at Karolinska Institutet consists of four senior scientists, six postdoctoral fellows, two graduate students a research engineer and two technicians.