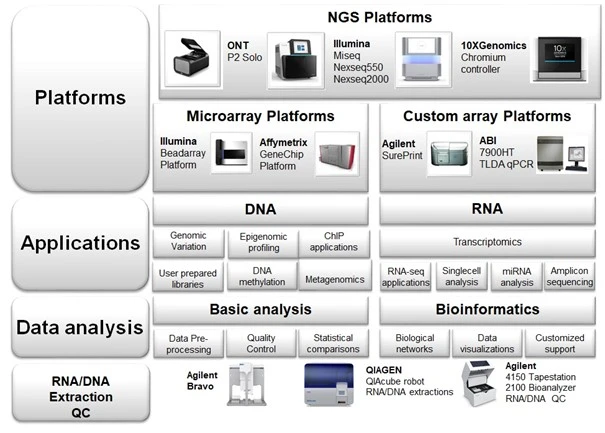
BEA - Applications
BEA provides and supports a number of different genomic applications. Please contact us to discuss your project.
RNA/DNA extractions and purifications
BEA offers service for isolation and purification of RNA and DNA from biological samples. For RNA extractions, we use different versions of the QIAgen RNeasy protocol which are designed for purification of total RNA from a wide variety of sources like animal cells, tissues, bacteria, and yeast in different formats. This includes RNA/DNA extractions from difficult samples like blood and FFPE.
Good results in downstream genomic analysis depend on the sampling and the quality of the DNA/RNA. Different samples demand different protocols and input buffers. Tissue samples can be homogenized with our Tissuelyzer prior to extraction.
Gene expression analysis
RNA sequencing is highly sensitive and accurate tool for measuring gene expression across the transcriptome. RNA sequencing allows detecting both known and novel features without prior knowledge. There are many different protocols available for preparing samples for RNA sequencing.
Below are some methods available at BEA:
- Whole transcriptome or Total RNA sequencing makes it possible to detect novel splice variants and analysis of both coding and non-coding RNA expression.
- Targeted RNA sequencing focus on specific transcripts of interest. Different selected library preparations by PCR or hybridization capture are available.
- mRNA sequencing is the most standardized and used method for analyzing gene expression. The method focuses on the coding transcripts and library preparation is performed by initial polyA selection from Total RNA.
- Small RNA profiling focuses on RNA sequences of <100 nt. Special protocols for capture and library preparation of desired fractions are thus needed.
Choosing the right library preparation method is crucial for the analysis outcome. Total cellular RNA is mainly composed of rRNA and is therefore often not of interest. For the purposes above we offer different library preparation services with or without depletion of rRNA. The Illumina stranded ligation protocol offers a streamlined, cost efficient, and scalable solution for transcriptome analysis. It is compatible with a wide range of RNA sequencing samples and targets different pools of RNA.
For small and pico amounts of RNA we offer transcriptome analysis using SMARTseq template-switching to capture full-length sequence information. Services include the Takara cDNA synthesis and library preparation by tagmentation.
Analysis by Microarrays
The core facility provides analysis services for the Affymetrix, Agilent and Illumina microarray platforms.
We have a long experience in expression analysis by microarrays and can offer extensive data analysis support and bioinformatics. Gene expression analysis is most often performed with the Affymetrix platform to investigate global gene expression as well as the transcriptional profiling in a wide range of organisms. Affymetrix provide four generations of expression arrays, Clariom D and S arrays, Transcriptome arrays (HTA) and 3' (IVT) arrays. Depending on the array type the amount of the required input amount varies from ~1ng-200ng. Each sample will be analyzed for its quality and quantity, amplified and terminally labeled before being hybridized following a standardized protocol.
The bioinformatic services include quality control, processing of raw data, comparisons of sample groups and annotations. The results will be summarized in Excel spreadsheets. The Affymetrix Clariom D arrays also allows exon-level analysis and alternative splicing analysis.
User prepared libraries/NGS sample submission
BEA is preparing libraries as described but also accepting libraries prepared by the user. Importantly, the submitted sequencing libraries has to meet requirements and important information regarding the sequencing has to be stated and submitted. Therefore, users shall always indicate type of flowcell, general sequencing setup, indexes and eventual custom primers that should be used during sequencing.
Information about library complexity and diversity are also important and should be communicated.
Library pools should be prepared in 10 nM concentration if possible. Libraries submitted to BEA has to be checked for concentration using fluorometric measurements (Qubit) and for quality Bioanalyzer (TapeStation).
Epigenetics and Methylation Analysis
BEA offers solutions for studies of gene regulation on the epigenetic level with the most commonly used applications for epigenetic studies.
Methods to determine the proportion of cytosine methylation in DNA can be performed at BEA. We offer microarray analysis with the Illumina Infinium Methylation EPIC Beadchips. Alternatively, whole genome bisulfite sequencing or RRBS (reduced representation bisulfite sequencing) which reduce the scale and cost of whole genome bisulfite sequencing can be performed. We also have service for EM-seq (enzymatic methylation sequencing) which is an enzyme-based DNA methylation sequencing technology that distinguishes between unmethylated cytosine and methylated 5-methylcytosine.
Chromatin immunoprecipitation sequencing, ChIP-seq, is a method for identifying protein interactions with DNA and differential histone modification sites across the genome. Other methods to study gene regulation and chromatin accessibility are ATAC seq (Assay for Transposase Accessible Chromatin), which gives information on the open chromatin regions in a genome and Cut and Tag sequencing which combines antibody-targeted controlled cleavage by a protein A-Tn5 fusion with DNA sequencing to identify the binding sites of DNA-associated proteins.
At BEA we offer service to construct ChIP sequencing libraries and sequencing of user provided ATAC and Cut and Tag sequencing libraries with complete downstream bioinformatics analysis.
Metagenomics
Metagenomic analysis can give insight into the interactions between microbials and help identify individual species within microbial habitats. With shotgun analysis, fragmented DNA extracted from environmental samples are sequenced to study not only the microbial species composition but also the gene functions and associated metabolic pathways. With 16S/18S, amplicon sequencing focused on sequencing specific target regions to understand the composition and diversity within a biological sample.
BEA offers library preparation service includes whole genome shotgun sequencing of microbiota samples and targeted 16s amplicon sequencing using the Zymo Quick 16s kit which targets different hypervariable regions of the 16s rRNA gene to help identify the presence of multiple bacterial species. The analysis can also be used in combination with Oxford Nanopore sequencing for targeting the full 16s region.
DNA sequencing and Genotyping
For analysis of DNA samples there is a broad range of library preparation methods and microarray based methods available for different purposes and applications. Choosing the right DNA sequencing method for your samples depends on the amount and quality of DNA, the organism and the goals of your study. Common genomic applications are Whole Genome and Exome sequencing which allows analysis of mutational variants of the genome and exome.
For smaller and more focused applications custom targeted sequencing is possible where designed panels of specific regions of the genome are targeted for sequencing. This includes different methods with gene panels, disease panels, or amplicon sequencing where custom regions or genes of interest are sequenced.
At BEA we have service to prepare your DNA samples for sequencing with different methods.
Appropriate library preparation protocols provided at BEA:
- NEB Next Ultra II DNA Library kit is designed for construction of high-quality libraries using a broad range of input amounts of DNA.
- Nextera DNA Library kits provide a fast and easy library prep workflow delivering whole-genome sequencing libraries.
- Nextera XT DNA Library kit prepares sequencing libraries for small genomes, PCR, amplicons, plasmids, metagenomics.
- Truseq DNA Nano and Truseq DNA PCR free reagents for whole genome sequencing applications including whole-genome resequencing and de novo assembly.
- Different options and versions of Illumina Infinium SNP Genotyping microarrays.
- Nanopore DNA sequencing of long fragments of DNA.
Long read sequencing
BEA has introduced long-read sequencing services with Oxford Nanopore to read long fragments of DNA or RNA for different applications to create more accurate genome assemblies, identifying large structural variations, phasing haplotypes, and characterizing epigenetic modifications. Please contact us for your specific needs.
Single cell sequencing service
BEA core facility provides service with the 10X Genomics Chromium platforms allowing solutions for single cell genomics, characterization and gene expression profiling. The Chromium controller device enables the analysis of large cell numbers at the highest capture efficiency. The technology allows for high-throughput single cell transcriptomics of many different cell types.
qPCR
qPCR is the standard for measuring changes in gene expression and can detect very small changes. Our quantitative real-time PCR ABI 7900HT enables high-throughput gene expression analysis with different chemistry. The most common approach for gene expression studies is a relative quantitation approach using reference genes for normalization that are not affected in the desired comparisons.
SYBR Green is a method based on intercalating nucleic acid staining dye while Taqman is a method based on hydrolysis probe. Both technologies are designed to generate fluorescence during the PCR, which allows real-time PCR machine to monitor the reaction in “real time”. BEA offers help to design your specific analysis. Please enquire about the specific primers and probes needed for your project.
BEA also offers service with predesigned 96 well plates and TLDA (Taqman low density arrays) cards which are 384 well micro fluidic cards preloaded with Taqman gene expression assays. You can customize the TLDA arrays with your genes of interest or choose among predesigned signatures arrays provided by Thermo.