Research theme: Biochemistry
Biochemical properties of TRP14 (TXNDC17)
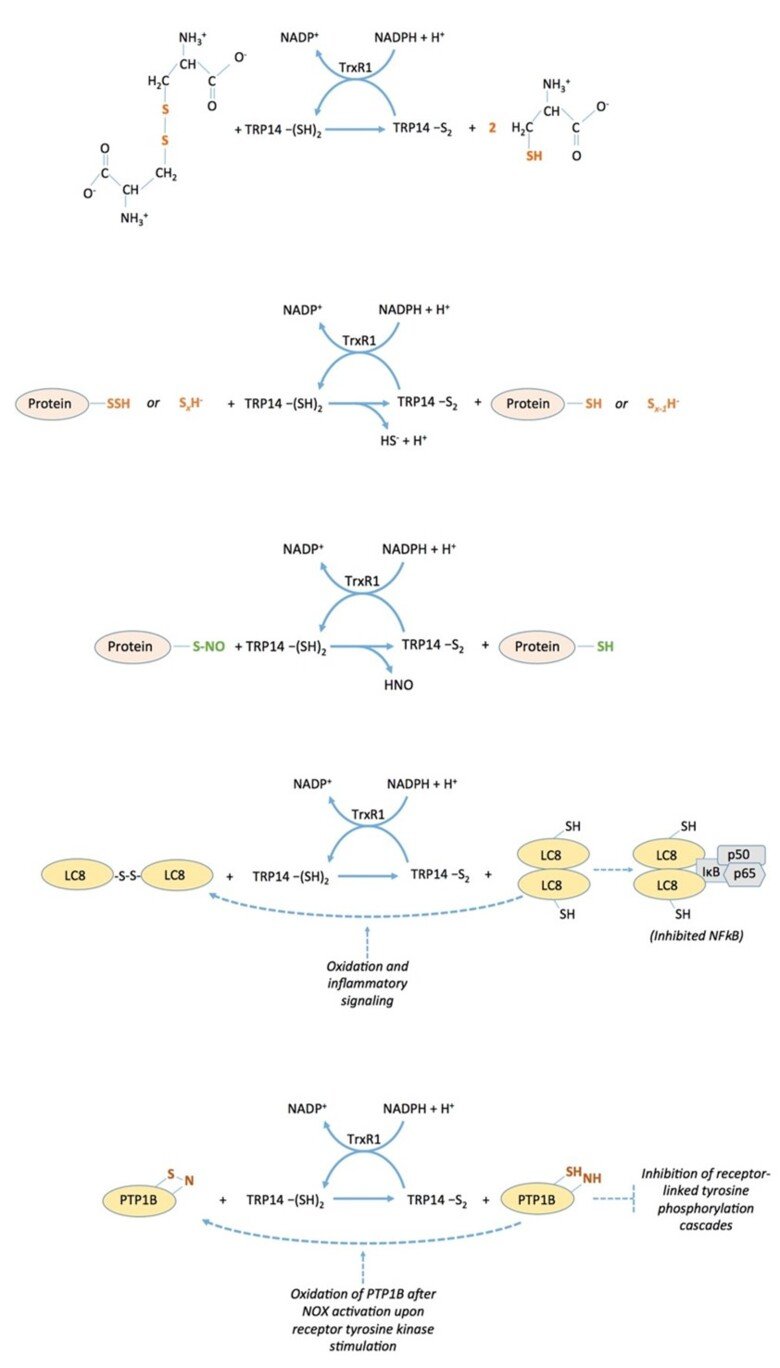
The thioredoxin-fold protein named TRP14 (thioredoxin Related Protein of 14 kDa), which is encoded by TXNDC17 in human (for thioredoxin domain-containing 17), is an interesting and little-studied protein that is ubiquitously expressed in cells in parallel with thioredoxin, a good substrate for thioredoxin reductase, but is not capable of reducing typical thioredoxin substrates such as ribonucleotide reductase, peroxiredoxins or methionine sulfoxide reductases. We found that TRP14 has a very good activity in reduction of cystine, persulfides and nitrosylated thiols, suggesting that it may be involved in diverse signaling events in cells. See “Thioredoxin-related protein of 14 kDa is an efficient L-cystine reductase and S-denitrosylase”, “A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems”, and “Thioredoxin-related protein of 14 kDa as a modulator of redox signalling pathways” for further information.
Redox regulation of intracellular signaling events
Regulation of PTP1B through the thioredoxin system
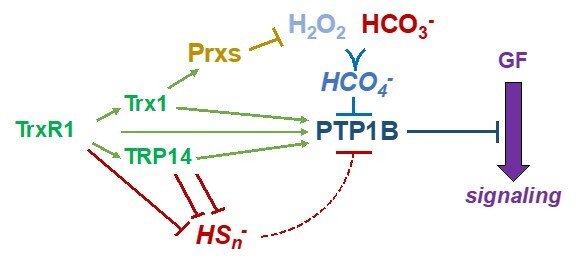
Protein tyrosine phosphatases (PTP enzymes) counteract tyrosine phosphorylation cascades triggered by tyrosine kinase receptors mediating the actions of growth factors such as PDGF, EGF or insulin. We have studied PTP1B at significant detail, showing how the thioredoxin system, with TrxR1, Trx1 with or without peroxiredoxins, as well as TRP14, efficiently acts to maintain PTP1B in a reduced and active state. These systems can also overcome inactivation of PTP1B by persulfidation triggered by polysulfides. A conundrum has thus been how PTP1B can be transiently oxidized and inactivated in conjunction with the oxidative burst from NADPH oxidases typically stimulated upon ligands binding to and activating tyrosine receptor kinases. Interestingly, we recently found that CO2/bicarbonate seems to be an obligate mediator of such PTP1B inactivation together with H2O2, likely by forming peroxymonocarbonate. This finding can have major implications with regards to the understanding of redox regulation and growth factor signaling in relation to acid-base balance under physiological as well as pathological conditions. For further information, see some of the following papers “Qualitative Differences in Protection of PTP1B Activity by the Reductive Trx1 or TRP14 Enzyme Systems upon Oxidative Challenges with Polysulfides or H2O2 Together with Bicarbonate”, “Bicarbonate is essential for protein-tyrosine phosphatase 1B (PTP1B) oxidation and cellular signaling through EGF-triggered phosphorylation cascades” and “Control of protein function through oxidation and reduction of persulfidated states”.
Regulation of PTPN22
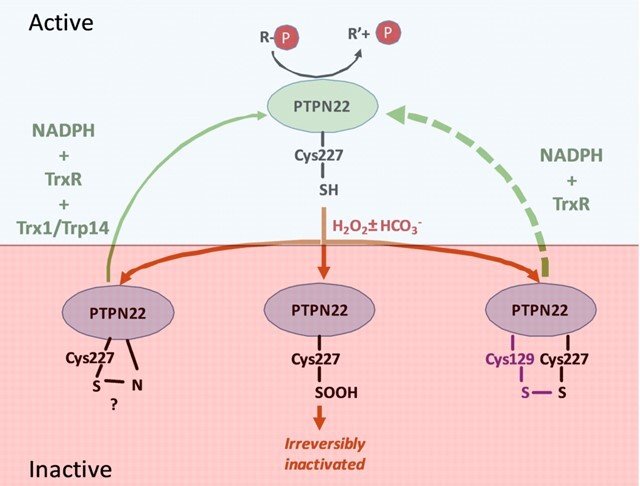
Together with the group of Prof. Rikard Holmdahl, we studied PTPN22, which is important in regulation of T cells and mutated in certain forms of autoimmunity. Similarly to PTP1B, we found that PTPN22 is also kept in a reduced and active state through the thioredoxin system, implying that dysregulation of the thioredoxin system in relation to PTPN22 function may also be involved in autoimmunity. For further details, see “Redox regulation of PTPN22 affects the severity of T-cell-dependent autoimmune inflammation”.
Regulation of STAT3 signaling
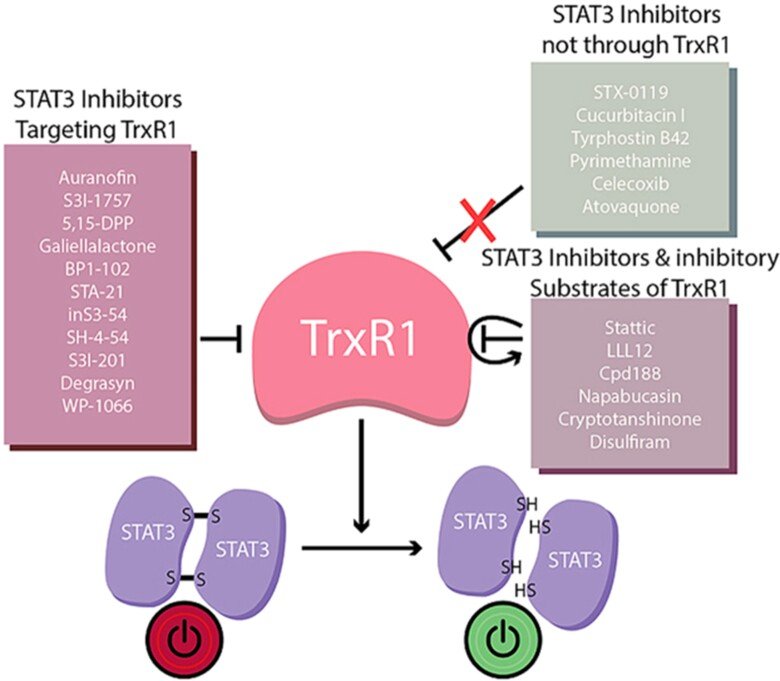
In collaborations with Dr. Page, we discovered that virtually all inhibitors of TrxR1 that we tested also inhibited activation of STAT3, an important transcription factor involved in cancer progression and regulation of antitumoral immunity. It had been known that STAT3 was redox sensitive, i.e. that oxidation of Cys residues in STAT3 into disulfide(s) would block its activation, but our findings clearly suggested that STAT3 function is fully TrxR1-dependent. Even more interesting, when we tested 23 small compounds previously claimed to be STAT3 inhibitors, 17 out of these were found to be TrxR1 inhibitor. These findings suggest that TrxR1 activity may be a critical factor for sustained STAT3 function and, conversely, that inhibitors of TRxR1 may inherently also be inhibitors of STAT3 activation. See ”Irreversible TrxR1 inhibitors block STAT3 activity and induce cancer cell death” and “To inhibit TrxR1 is to inactivate STAT3-Inhibition of TrxR1 enzymatic function by STAT3 small molecule inhibitors” for further information.
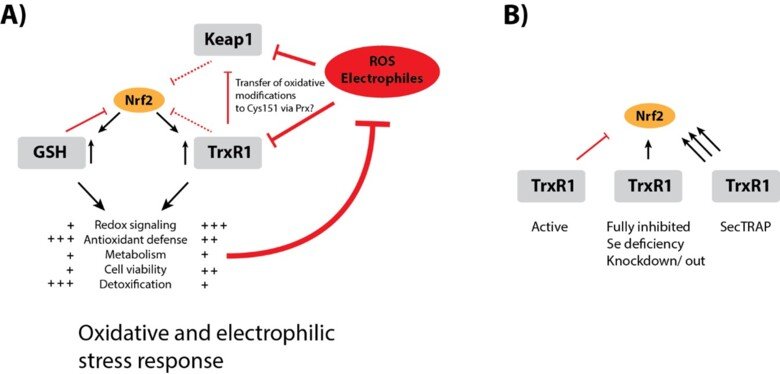
Regulation of Nrf2 signaling
The Nrf2/Keap-1 system is key for regulating redox homeostasis and metabolism, with both thioredoxin and thioredoxin reductase also being Nrf2 targets. Interestingly, most inhibitors of TrxR1, or conditional knockout of the enzyme in mouse tissues, typically leads to strong upregulation of Nrf2 – also without evident signs of oxidative stress. Could it be that TrxR1 is a negative regulator of Nrf2 activation, by yet unknown mechanism(s)? This is a possibility discussed in a review of ours, which also cites the many observations suggesting this could be the case. See “TrxR1 as a potent regulator of the Nrf2-Keap1 response system” and therein cited publications for further information.
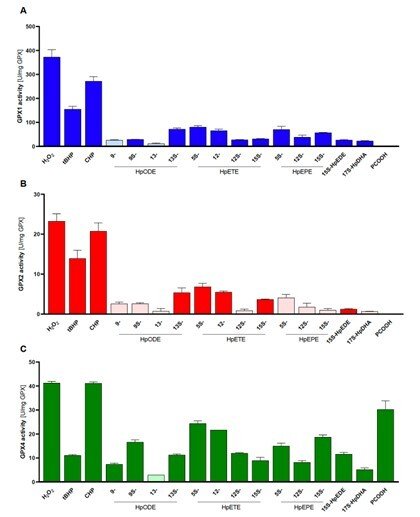
Side-by-side comparisons of GPX1, GPX2 and GPX4
In a collaboration with Prof. Anna Kipp and her group in Jena, Germany, we compared the substrate specificities of human glutathione peroxidases GP1, GPX2 and GPX4 with pure enzyme systems and identical conditions. This had never been done before because it has been very difficult and laborious to obtain the pure enzymes, but this study was now enabled by our recent method development to produce recombinant selenoproteins with internal Sec residues, such as the GPX isoenzymes. In this study, we found that GPX1 and GPX2 have very similar substrate specificities, but with GPX1 having much higher turnover. GPX4 could also be confirmed as the only out of the three isoforms that efficiently reduce lipid hydroperoxide substrates, while all three isoforms can easily reduce H2O2. See “Side-by-side comparison of recombinant human glutathione peroxidases identifies overlapping substrate specificities for soluble hydroperoxides” for further information.
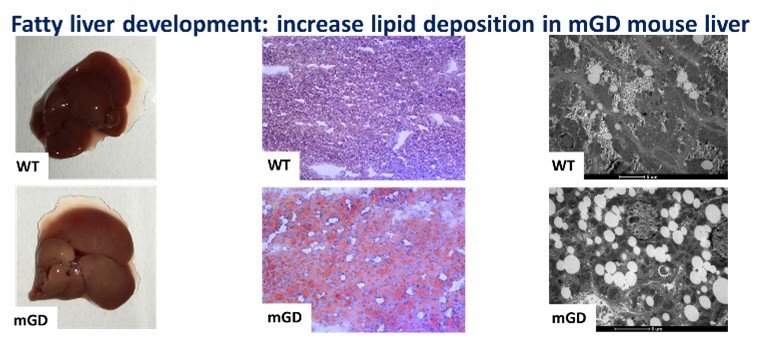
Role of mitochondrial Grx2 in lipid metabolism
The knowledge of liver biology is continuously growing, and we believe that the redox regulation of specific liver pathways is an aspect in which major advances can be reached. Understanding that redox enzymes such as Glutaredoxin 2, Grx2, can have an impact in mitochondrial function and, as we showed in our recent paper “Mitochondrial depletion of glutaredoxin 2 induces metabolic dysfunction-associated fatty liver disease in mice”, on lipid accumulation and glycogen storage, could represent a key point that may shed light on the pathophysiology of different liver diseases and translate this in better clinical practices. The mitochondrial isoform of glutaredoxin (Grx2) is a glutathione-dependent oxidoreductase that is involved in glutathionylation/de-glutathionylation of proteins. In order to investigate the role of mitochondrial Grx2, a unique mouse model specifically depleted of Grx2 in mitochondria (mGD) was employed. This condition unexpectedly determined a series of metabolic imbalances especially in lipid metabolism, bringing to a phenotype attributable to the nonalcoholic fatty liver disease.
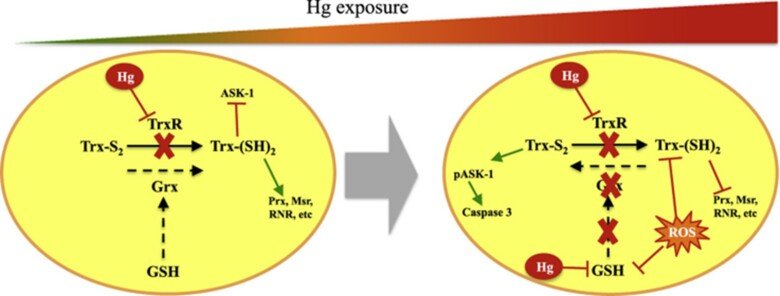
Role of glutaredoxin and thioredoxin systems in mercury toxicity
Mercury (Hg) compounds are well-established neurotoxicants. The ability to cross the blood–brain barrier is a key feature explaining the tropism of these compounds to the central nervous system where they accumulate and once a threshold is reached, toxicity will ensue, characterized by motor and cognitive function deficits which are especially pernicious during in utero development. We have studied the role of redox pathways in mercury toxicity at significant detail, showing interactions between mercury compounds and the glutathione and thioredoxin systems in neuronal cells, and we have characterized how inhibition of the Trx system by mercurials can regulate cell death.
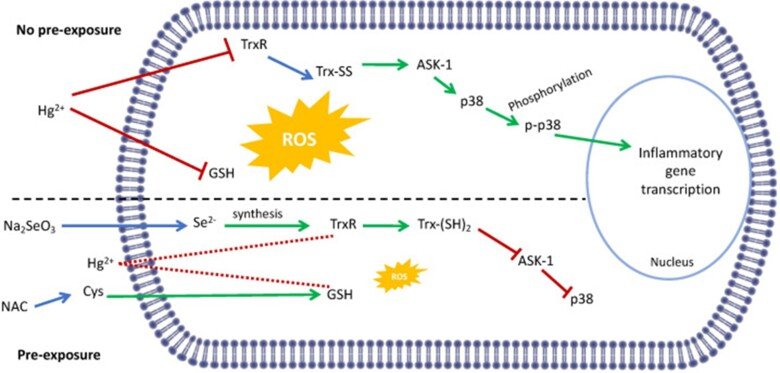
Regarding the immunotoxic effects of Hg we showed that disruption of redox systems by Hg2+ prompts the activation of p38 leading to transcription of pro-inflammatory genes in microglia cells. Treatment with a cysteine precursor or sodium selenite, which caused an increase in basal GSH and TrxR levels, respectively, prevented the activation of p38 and the transcription of pro-inflammatory cytokines. For further information, see the following papers “N-Acetylcysteine or Sodium Selenite Prevent the p38-Mediated Production of Proinflammatory Cytokines by Microglia during Exposure to Mercury (II)” and “Impaired cross-talk between the thioredoxin and glutathione systems is related to ASK-1 mediated apoptosis in neuronal cells exposed to mercury”.
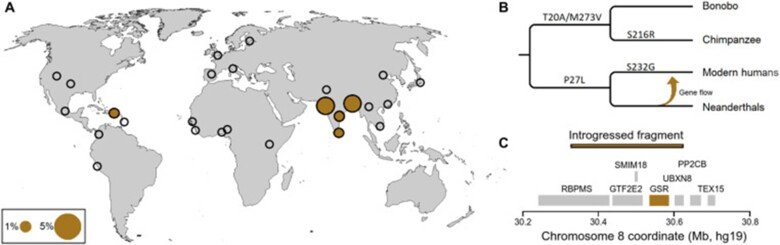
Comparison of Neanderthal and modern human glutathione reductases
Some ancestral genetic variants and some genetic variants unique to Neanderthals exist among present-day people. There are around one hundred protein-encoding genes that have mutational changes differing between Neanderthals and modern humans that are carried by almost all living people today. One of these proteins is glutathione reductase which is part of the body's defense against oxidative stress. We characterized in detail the present-day versus the Neanderthal protein and we demonstrated that the latter one produced more reactive oxygen radicals in an NADPH dependent reaction. This is the third protein change unique to present-day humans that has been studied so far. We show that people who carry the Neanderthal protein have a higher risk of developing vascular disease and inflammatory bowel disease, two diseases that are linked to oxidative stress. For more details see “A substitution in the glutathione reductase lowers electron leakage and inflammation in modern humans”.
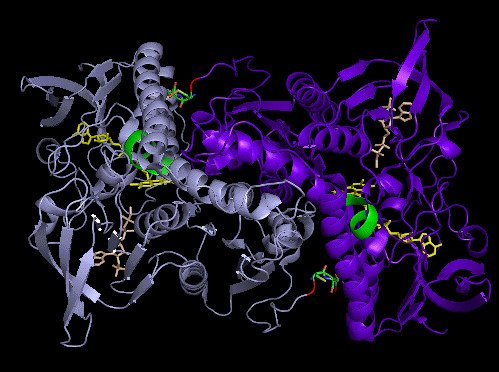
Crystal structure and catalysis of selenoprotein TrxR1
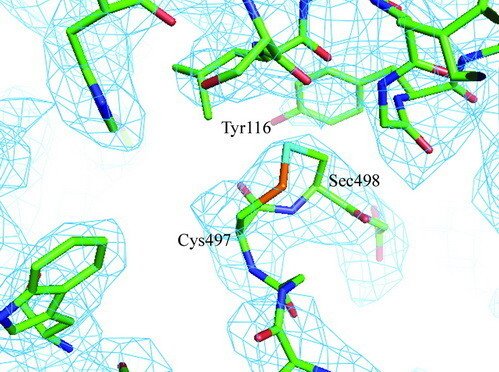
Selenoproteins contain a highly reactive 21st amino acid selenocysteine (Sec) encoded by recoding of a predefined UGA codon. Because of a lack of selenoprotein supply, high chemical reactivity of Sec, and intricate translation machineries, selenoprotein crystal structures are difficult to obtain. Structural prerequisites for Sec involvement in enzyme catalysis are therefore sparsely known.
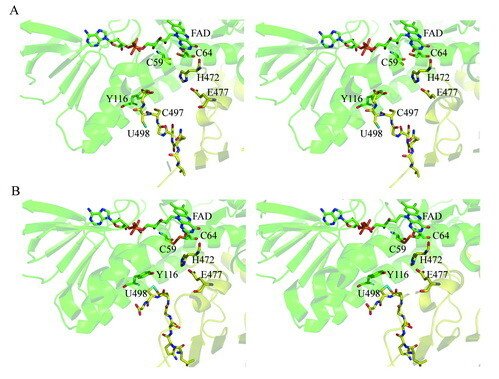
Here we present the crystal structure of catalytically active rat thioredoxin reductase 1 (TrxR1), revealing surprises at the C-terminal Sec-containing active site in view of previous literature. The oxidized enzyme presents a selenenylsulfide motif in trans-configuration, with the selenium atom of Sec-498 positioned beneath the side chain of Tyr-116, thereby located far from the redox active moieties proposed to be involved in electron transport to the Sec-containing active site. Upon reduction to a selenolthiol motif, the Sec residue moved toward solvent exposure, consistent with its presumed role in reduction of TrxR1 substrates or as target of electrophilic agents inhibiting the enzyme.
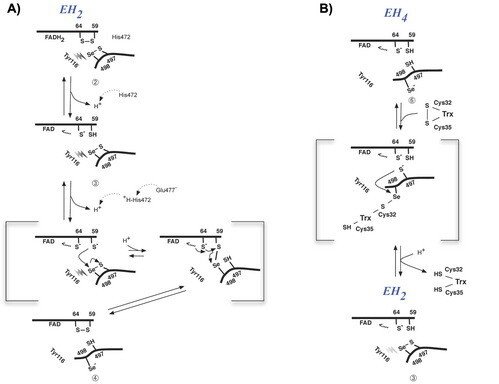
The prerequisites for this model postulates the following: Cys-64 makes the FAD-charge-transfer complex; Cys-59 reduces the selenenylsulfide by an intermediary selenenylsulfide interchange with Sec-498 with His-472 and Glu-477 potentially acting as proton acceptors (in two successive steps, through His-472); Trx becomes reduced through an intermediate selenenylsulfide between Cys-32 of Trx and Sec-498 of TrxR, and Tyr-116 acting by (i) facilitating selenenylsulfide reduction in the reductive half-reaction through resonance effects and, possibly, (ii) facilitating release of the intermediate selenenylsulfide with Trx by its interaction with the selenenylsulfide between Cys-497 and Sec-498. A, details of the reductive half-reaction. This model is in agreement with data in the literature and with modeling based upon the crystal structure, postulating that an intermediate selenenylsulfide between Cys-59 and Sec-498 is formed. Note, however, that formation of an intermediate disulfide between Cys-59 and Cys-497 cannot be excluded as an alternative pathway for the reductive half-reaction. B, details of a model for Trx reduction. This model postulates that Trx makes a mixed selenenylsulfide between Cys-32 and Sec-498, whereupon a thiolate is formed at Cys-497 (proton transferred to Cys-35 of Trx), which attacks the mixed selenenylsulfide, thereby enabling formation of the selenenylsulfide at the C terminus of TrxR1; this motif is subsequently interacting with Tyr-116. Further reading in "Crystal structure and catalysis of the selenoprotein thioredoxin reductase 1".
Redox systems of parasites
Selenoprotein thioredoxin reductases of filarial parasites
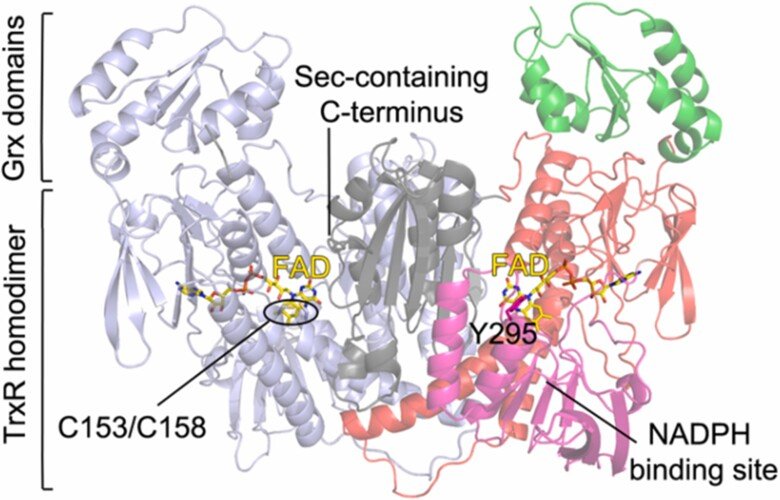
Together with the groups of Drs. David Williams and Francesco Angelucci we characterized the thioredoxin reductase (TrxR) selenoproteins from Brugia malayi and Onchocerca volvulus, which are filarial nematode parasites that can cause lymphatic filariasis and onchocerciasis, respectively. We found both enzymes to have a domain organisation resembling that of human TGR (thioredoxin glutathione reductase) having an additional N-terminal glutaredoxin-domain added to the typical selenoprotein TrxR domain. Interestingly, however, the parasite variants of this enzyme did not possess glutathione reductase activities and, thus, the Grx domains have yet unknown function. The specific architecture of these enzymes might, potentially, open the way for specific therapies against these parasites, provided that specific inhibitors could be developed. For further information, see “Biochemical and structural characterizations of thioredoxin reductase selenoproteins of the parasitic filarial nematodes Brugia malayi and Onchocerca volvulus”. For a general review on parasite TrxR variants, as well as human TrxR selenoproteins, and the possibility of targeting these for treatment of disease, see “Thioredoxin reductase selenoproteins from different organisms as potential drug targets for treatment of human diseases”.
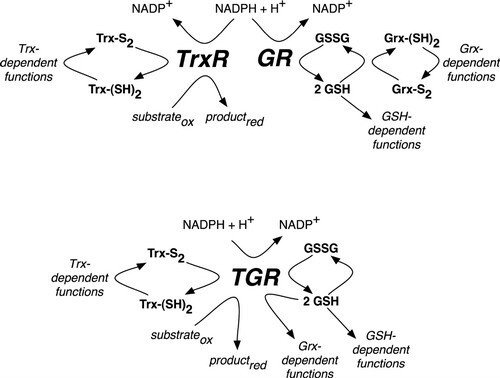
Redox Pathways in Schistosoma mansoni
In mammals (upper pathway), electrons from NADPH are transferred to an oxidoreductase flavoenzyme, either thioredoxin reductase (TrxR) or glutathione reductase (GR). Electrons are then transferred from the oxidoreductase flavoenzyme to the appropriate electron carrier, either oxidized thioredoxin (Trx-S2) or glutathione disulfide (GSSG) converting them to reduced thioredoxin (Trx-[SH]2) or glutathione (GSH), respectively. Trx-(SH)2 and GSH then supply reducing equivalents for a number of different reactions, including those that are glutaredoxin (Grx)-dependent. In S. mansoni (lower pathway), TrxR and GR are replaced with a unique oxidoreductase flavoenzyme, TGR, which provides reducing equivalents for Trx-, GSH- and Grx-dependent reactions. Further reading in "Thioredoxin glutathione reductase from Schistosoma mansoni: an essential parasite enzyme and a key drug target".
Those original findings became the basis for additional studies of potential inhibitors of the selenoprotein from Schistosoma, which may work as drug leads for the development of novel therapies against this pathogenic parasite, as can be read more about in “Fragment-Based Discovery of a Regulatory Site in Thioredoxin Glutathione Reductase Acting as "Doorstop" for NADPH Entry”, “Structure mechanism insights and the role of nitric oxide donation guide the development of oxadiazole-2-oxides as therapeutic agents against schistosomiasis” or “Characterization of Lead Compounds Targeting the Selenoprotein Thioredoxin Glutathione Reductase for Treatment of Schistosomiasis”.
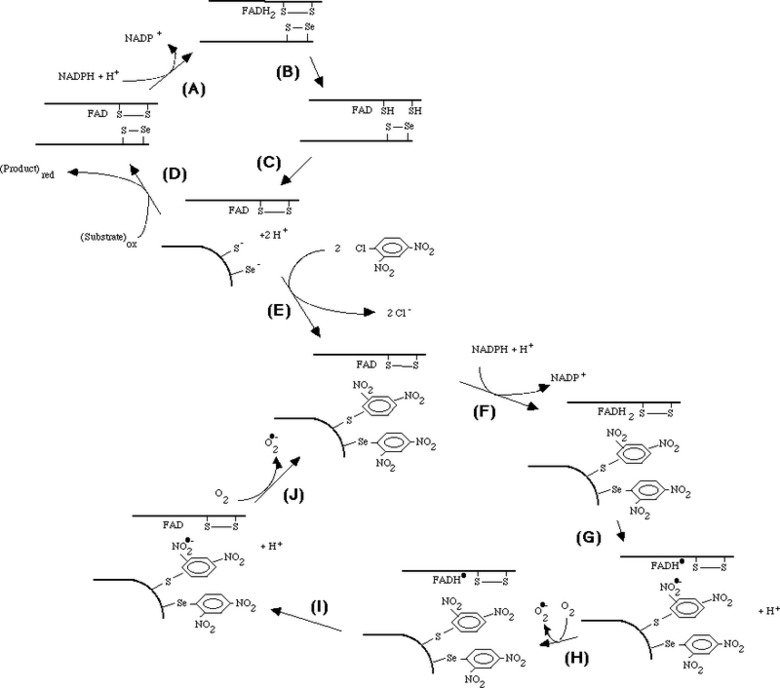
Mechanism of irreversible inactivation and concomitant induction of an NADPH oxidase activity upon derivatization of TrxR1 with DNCB
Mechanism of irreversible inactivation and concomitant induction of an NADPH oxidase activity upon derivatization of TrxR1 with DNCB. 1-chloro-2,4-dinitrobenzene (DCNB, CDNB) was found to irreversibly inactivate reduced TrxR with a high affinity and specificity. This reaction gave a concomitant induction of an NADPH oxidase activity by the derivatized enzyme, as shown in "1-Chloro-2,4-dinitrobenzene is an irreversible inhibitor of human thioredoxin reductase. Loss of thioredoxin disulfide reductase activity is accompanied by a large increase in NADPH oxidase activity".
Later knowing the selenoprotein nature and primary structure of TrxR1 we also showed that the selenocysteine-containing motif in fact was the primary target for dinitrophenyl-derivatization and that the NADPH oxidase activity produced superoxide. This was presented in "Mammalian thioredoxin reductase is irreversibly inhibited by dinitrohalobenzenes by alkylation of both the redox active selenocysteine and its neighboring cysteine residue".
Taken together, the mechanistic model shown in the figure could be formulated. The model involves reduction of the C-terminal selenenylsulfide by NADPH (A - C), which then becomes a target for DNCB alkylation (E) explaining the irreversible inhibition prohibiting the normal cycle of reduction of substrates (D). The FAD of the dnp-derivatized enzyme can still be reduced by NADPH (F) but is proposed to participate in two consecutive one-electron transfers to the nitro groups of the dnp moieties, producing nitro anion radicals (G, I) which may react with molecular oxygen to form superoxide (H, J). The FAD may again be reduced by NADPH (F), thereby forming the NADPH oxidase activity observed experimentally. The model is further discussed in "Superoxide production by dinitrophenyl-derivatized thioredoxin reductase--a model for the mechanism and correlation to immunostimulation by dinitrohalobenzenes".
We also investigated the cytotoxicity and caspase activation of both DNCB and other nitroaromatic compounds that we identified as new inhibitors of TrxR1. Specific inhibition of cellular TrxR1 over glutathione reductase correlated well with caspase activation and cytotoxicity in both lung and cervix carcinoma cell lines. This was a collaboration with Dr. Narimantas Cenas and coworkers, Vilnius, Lithuania, and the findings are described in "Interactions of nitroaromatic compounds with the mammalian selenoprotein thioredoxin reductase and the relation to induction of apoptosis in human cancer cells".
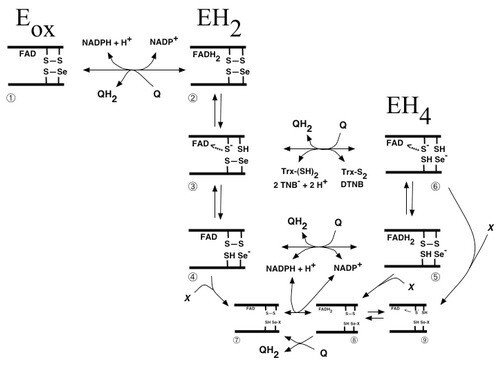
Scheme of quinone metabolism by mammalian TrxR
Many quinone compounds may interact with TrxR, either as substrates in one- as well as two-electron transfer reactions or as inhibitors, either competitively or irreversibly. This figure schematically summarizes the three major redox states of mammalian TrxR (oxidized enzyme, two-electron reduced or four-electron reduced species), with the selenolthiol motif of species nr 6 reducing any of the many substrates of this enzyme. Shown is also the reduction of quinones directly by the reduced flavin moiety in species 2 or 5 of the unmodified enzyme, or species 8 of the enzyme irreversibly inhibited for its normal reactions by derivatisation of the selenolate residue in by an electrophilic compound (“X”), which may also be a quinone derivative. This was a collaboration with Dr. Narimantas Cenas and coworkers, Vilnius, Lithuania, and the figure was taken from "Interactions of quinones with thioredoxin reductase: a challenge to the antioxidant role of the mammalian selenoprotein".
Later studies have shown that TrxR1 interacts with many different quinone compounds, in a highly intricate manner. This includes its reduction of, but also crosslinking by, benzoquinone, as shown in “Inhibition and crosslinking of the selenoprotein thioredoxin reductase-1 by p-benzoquinone”, as well as the interactions with pyrroloquinoline quinone (PQQ) that is a redox active cofactor for bacterial quinoproteins, which was found to stimulate the redox cycling of TrxR1 with a third quinone, juglone (see “Pyrroloquinoline quinone modulates the kinetic parameters of the mammalian selenoprotein thioredoxin reductase 1 and is an inhibitor of glutathione reductase”). With regards to the interactions of TrxR1 with juglone (walnut toxin), which is a defining characteristics of SecTRAPs (see https://ki.se/en/mbb/thioredoxin-reductase-1-in-relation-to-cancer), we discovered that the C-terminal Cys residue of TrxR1, neighboring the Sec residue, is of major importance. This further illustrated how intricate the biochemistry of TrxR1 is, particularly but not exclusively in its reactions with quinone compounds. See “Details in the catalytic mechanism of mammalian thioredoxin reductase 1 revealed using point mutations and juglone-coupled enzyme activities” for further discussions on that topic.